Postać kliniczna rdzeniowego zaniku mięśni może być różna w zależności od wieku wystąpienia objawów i stopnia nasilenia choroby, ale częstymi objawami lub oznakami są hipotonia (zespół wiotkiego dziecka) i/lub osłabienie oraz zanik mięśni2,3: |
|
|
|
Charakterystyka rdzeniowego zaniku mięśni
0–6 MIESIĘCY (postać niemowlęca)1,2,4
Najwyższy poziom rozwoju motorycznego
NIEZDOLNOŚĆ DO SAMODZIELNEGO SIEDZENIA
(„pacjenci niesiedzący”)
Długość życia
PONIŻEJ 2 LAT
Fenotyp
TYP I
(znany również jako choroba Werdinga-Hoffmanna)
Objawy kliniczne
- Hipotonia i zaburzenia kontrolowania ruchów głowy
- Leżenie na wzak w pozycji żaby
- Słaby płacz
- Słaby kaszel
- Połykanie, karmienie i radzenie sobie z wydzieliną powstającą w jamie ustnej zostają zaburzone przed ukończeniem 1. roku życia
- Atrofia i fascykulacje języka
- Osłabienia mięśni i hipotonia w obrębie kończyn i tułowia
- Osłabienie mięśni międzyżebrowych (uwaga, choroba nie atakuje początkowo przepony)
- Oddech paradoksalny
- Tułów przypominający kształtem dzwon z zapadniętą ścianą klatki piersiowej i protruzją jamy brzusznej
Aby dowiedzieć się, jak różne aspekty opieki wiążą się
z poszczególnymi objawami przedmiotowymi i podmiotowymi SMA, kliknij tutaj.
7–18 MIESIĘCY (postać pośrednia)2,4-6
Najwyższy poziom rozwoju motorycznego
ZDOLNOŚĆ DO SAMODZIELNEGO SIEDZENIA („pacjenci siedzący”)
Długość życia
>2 LATA
70% DOŻYWA 25 ROKU ŻYCIA
Fenotyp
TYP II
(znany również jako choroba Dubowitza)
Objawy kliniczne
- Porażenie opuszkowe z trudnościami w połykaniu, które może prowadzić do słabych przyrostów masy ciała
- Słabe mięśnie międzyżebrowe
- Oddychanie przeponowe
- Problemy z kaszlem i usuwaniem wydzieliny z tchawicy
- Łagodne drgawki podczas prostowania palców lub podczas prób chwytania
- Kifoskolioza lub skolioza wymagająca stabilizowania lub operacji kręgosłupa
- Przykurcze stawów
Aby dowiedzieć się, jak różne aspekty opieki wiążą się
z poszczególnymi objawami przedmiotowymi i podmiotowymi SMA, kliknij tutaj.
PONAD 18 MIESIĘCY (postać dziecięca)1,2,7
Najwyższy poziom rozwoju motorycznego
ZDOLNOŚĆ SAMODZIELNEGO CHODZENIA (możliwa stopniowa utrata tej zdolności)
Długość życia
STATYSTYCZNA
Fenotyp
TYP III
(znany również jako choroba Kugelberga-Welander)
Objawy kliniczne
- Skolioza
- Problemy z przełykaniem
- Kaszel, hipowentylacja w trakcie snu
- Bóle mięśni
- Objawy przeciążenia stawów
Aby dowiedzieć się, jak różne aspekty opieki wiążą się
z poszczególnymi oznakami i objawami SMA, kliknij tutaj.
PÓŹNY OKRES DOJRZEWANIA/ DOROSŁOŚĆ (postać dorosła)1,2,4
Najwyższy poziom rozwoju motorycznego
PEŁNY
Długość życia
STATYSTYCZNA
Fenotyp
TYP IV
Objawy kliniczne
- Objawy fizykalne są podobne do dziecięcej postaci SMA, ze stopniowo pojawiającymi się objawami polegającymi na osłabieniu, drgawkach i skurczach mięśni występującymi po raz pierwszy pod koniec okresu dojrzewania lub w dorosłości.
Aby dowiedzieć się, jak różne aspekty opieki wiążą się
z poszczególnymi objawami przedmiotowymi i podmiotowymi SMA, kliknij tutaj.
DHMN* (postać młodzieńcza)8–10
Najwyższy poziom rozwoju motorycznego
PEŁNY
Długość życia
STATYSTYCZNA
Fenotyp
TYP V
Objawy kliniczne
- Osłabienie i zanik mięśni pojawiające się najpierw w kończynach górnych
- Ostatecznie u połowy pacjentów pojawia się osłabienie kończyn dolnych
Aby dowiedzieć się, jak różne aspekty opieki wiążą się
z poszczególnymi oznakami i objawami SMA, kliknij tutaj.
Uwaga: W artykułach internetowych i badaniach klinicznych opisane objawy rdzeniowego zaniku mięśni są częściej grupowane według „typu” (I–V).
Naturalny przebieg rdzeniowego zaniku mięśni charakteryzuje się postępującym zanikiem mięśni i stopniową utratą funkcji ruchowych.5,6,8
Opracowano szereg skal funkcji ruchowych, które są przydatne do oceny przebiegu rdzeniowego zaniku mięśni oraz reakcji na eksperymentalne środki lecznicze w badaniach klinicznych.11–13
Przykłady obejmują†:
Skala do oceny sprawności nerwowo-mięśniowej niemowląt opracowana przez Szpital Dziecięcy w Filadelfii (CHOP INTEND) jest wykorzystywana do oceny zdolności motorycznych niemowląt z rdzeniowym zanikiem mięśni11,14: |
|
|
|
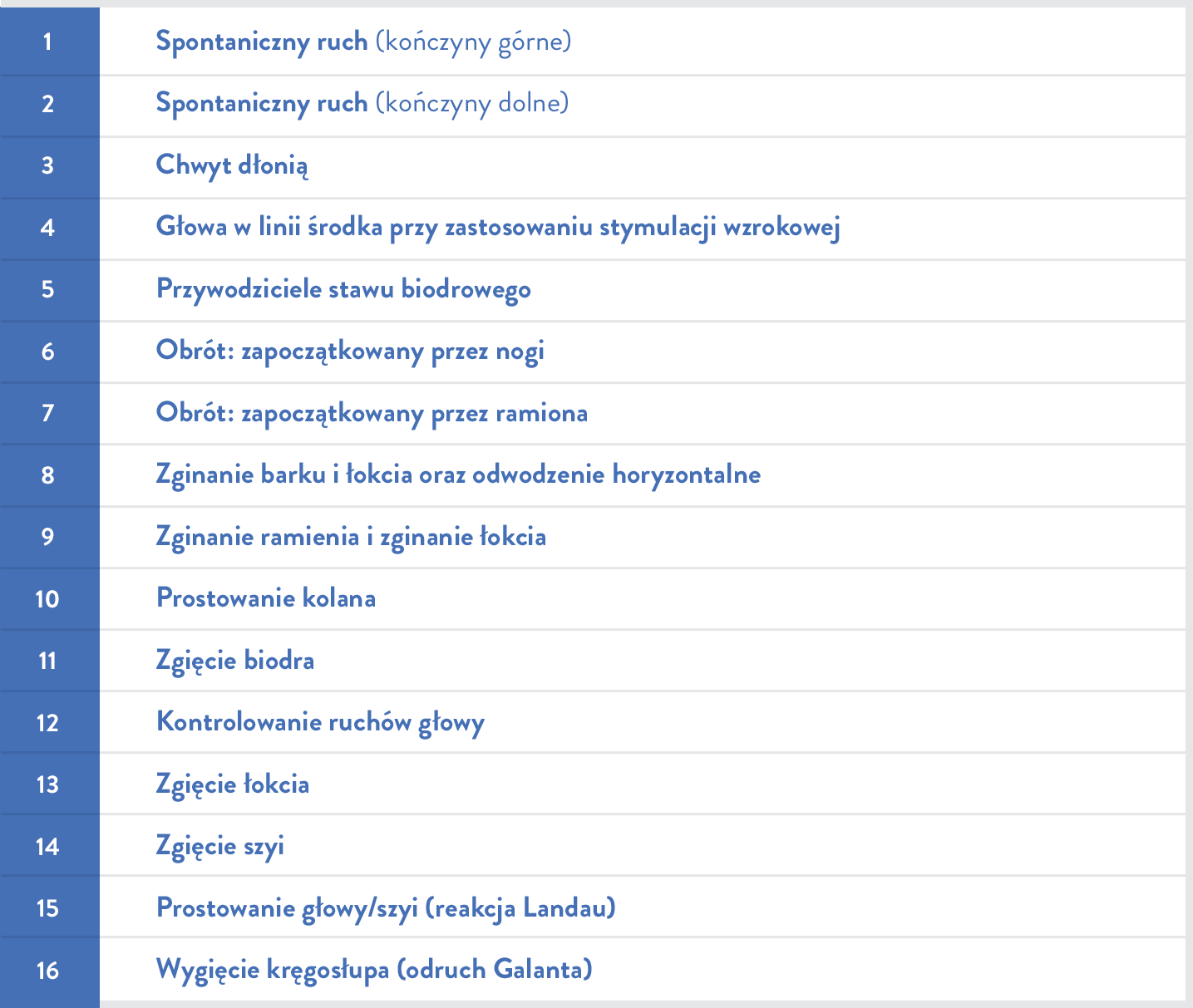
|
Skala HINE (Hammersmith Infant Neurological Examination) jest narzędziem do oceny funkcji ruchowych będącym prostą metodą punktacji zdolności motorycznych u dzieci w wieku od 2 miesięcy do 2 lat12,15: |
|
|
|
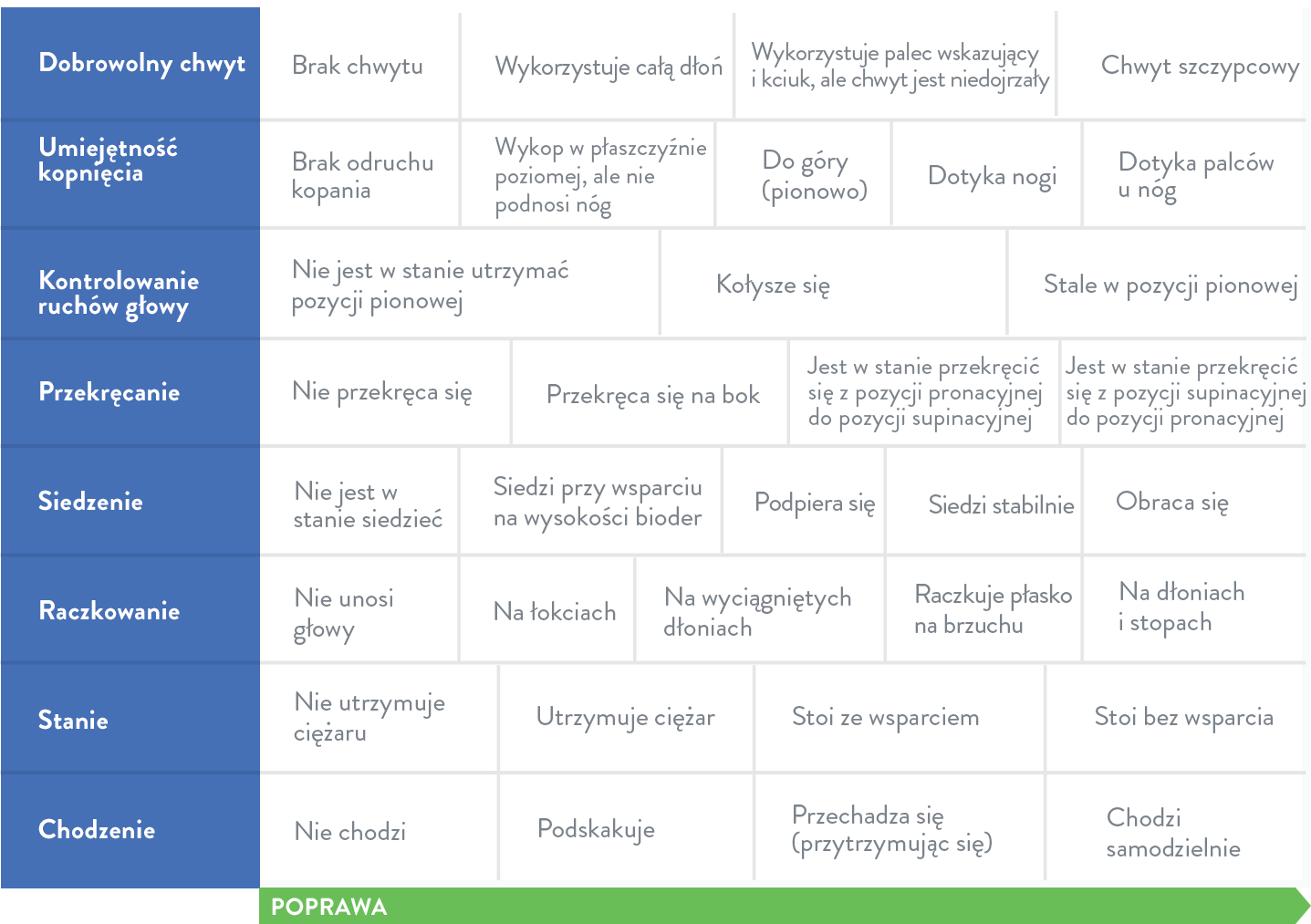
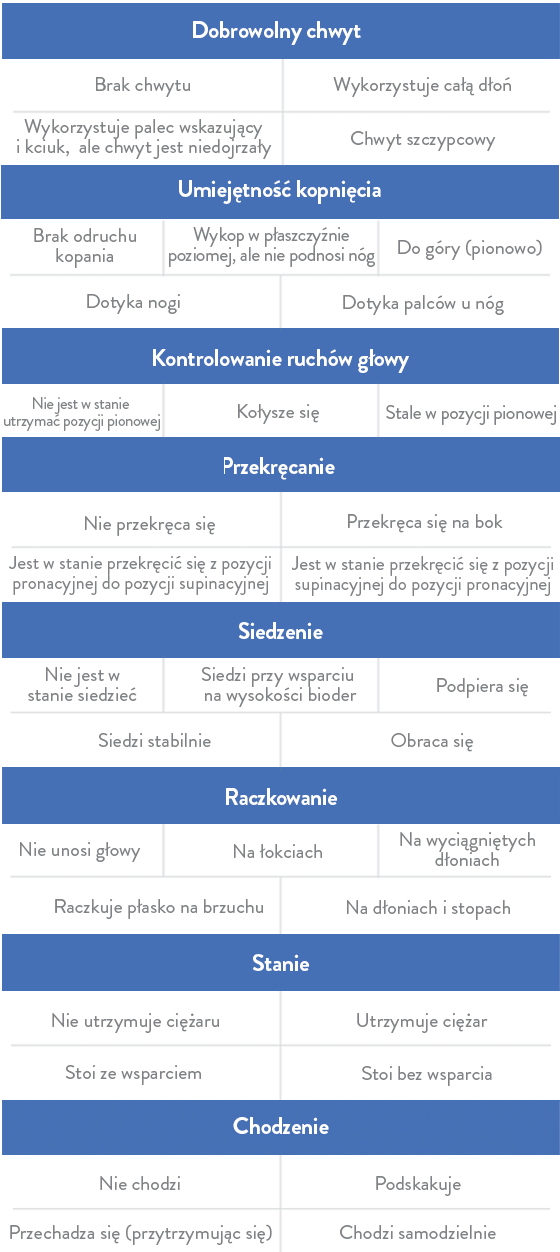
|
Skala HFMSE (Hammersmith Functional Motor Scale — Expanded) jest skalą zweryfikowaną, zatwierdzoną, wykorzystywaną w kilku badaniach klinicznych do oceny funkcji ruchowych dzieci chorych na rdzeniowy zanik mięśni. Do HFMSE dodano 13 istotnych klinicznie elementów pochodzących ze skali GMFM (Gross Motor Function Measure) dotyczących czynności leżenia/przewracania się, raczkowania, raczkowania/klękania, wstawania i chodzenia/biegania/skakania13,16: |
|
|
|
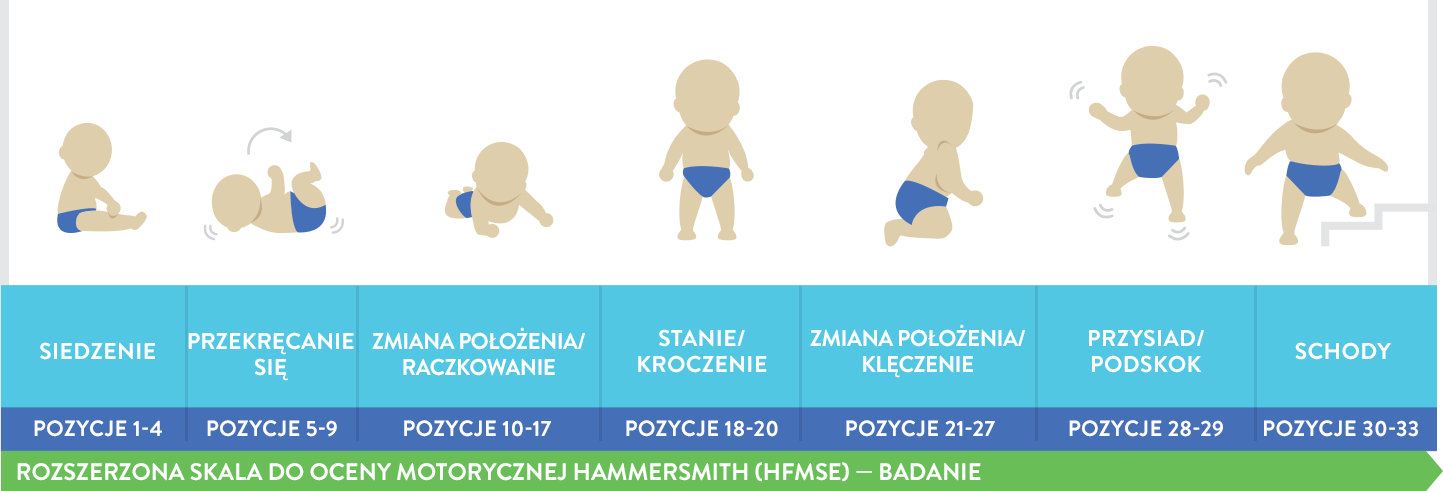
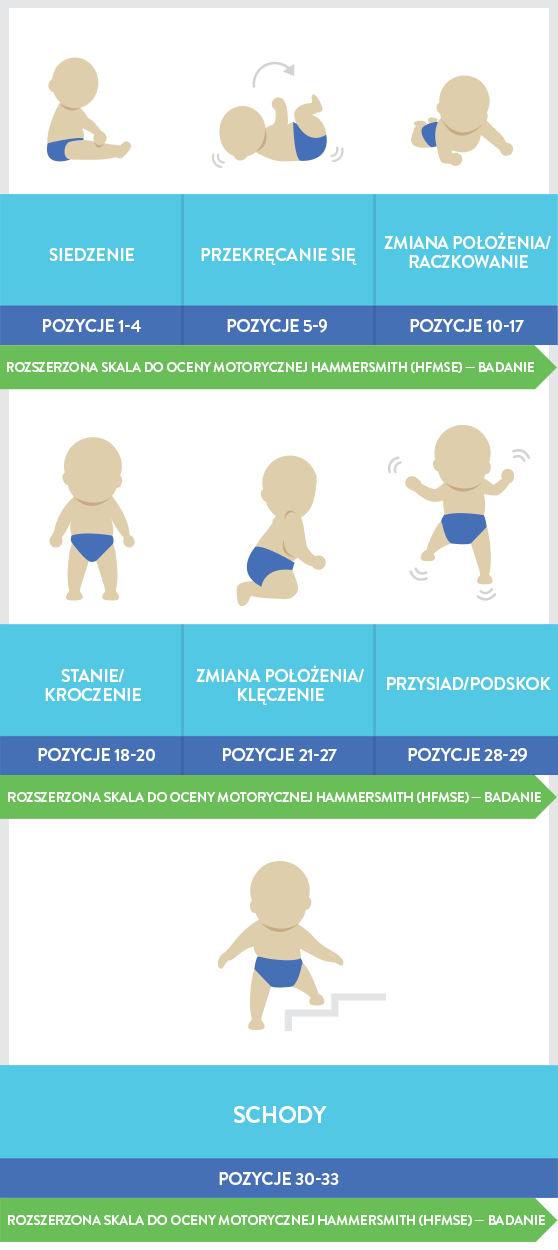
|
†Nie jest to wyczerpująca lista skali funkcji ruchowych.
W przypadku rdzeniowego zaniku mięśni, do oceny stanu neuronów ruchowych można zastosować badania elektrofizjologiczne.17
- Badanie złożonego potencjału czynnościowego (CMAP) jest techniką polegającą na mierzeniu elektrofizjologicznej reakcji mięśnia lub grupy mięśni po podrażnieniu nerwu obwodowego.18
- Ocena liczby jednostek motorycznych (MUNE) jest metodą, w której ocenia się liczbę jednostek motorycznych unerwiających dany mięsień. Wyniki metody MUNE oblicza się na podstawie stosunku maksymalnego złożonego potencjału z mięśnia (CMAP) i potencjału pojedynczego neuronu ruchowego (SMUP).
- Wartość SMUP jest mierzona poprzez przykładanie elektrody stymulującej do wielu punktów na całej długości nerwu ruchowego.19
U niektórych osób chorych na rdzeniowy zanik mięśni wartość CMAP może gwałtownie spadać.17
Spadki CMAP są związane z wystąpieniem objawów u dzieci cierpiących na niemowlęcą postać (typ I) rdzeniowego zaniku mięśni.17

PIŚMIENNICTWO
1. Prior TW, Russman BS. Spinal muscular atrophy. Strona internetowa NCBI Bookshelf. http://www.ncbi.nlm.nih.gov/books/NBK1352/?report=printable. Ostatnia aktualizacja: 14 listopada 2013 r. Data udostępnienia: 15 kwietnia 2016 r. 2. Wang CH, Finkel RS, Bertini ES, et al; and Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027–1049. 3. MedlinePlus. Medical Encyclopedia. https://www.nlm.nih.gov/medlineplus/encyclopedia.html. Ostatnia aktualizacja: 21 kwietnia 2016 r. Data udostępnienia: 25 kwietnia 2016 r. 4. Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46(1):1–12. 5. Darras BT, Royden Jones H Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician’s Approach. 2nd ed. London, UK: Elsevier; 2015. 6. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–2133. 7. Online Mendelian Inheritance in Man. Spinal muscular atrophy, Type III; SMA3. http://www.omim.org/entry/253400. Ostatnia aktualizacja: 7 lutego 2013 r. Data udostępnienia: 26 kwietnia 2016 r. 8. Genetics Home Reference. SMN1. https://ghr.nlm.nih.gov/gene/SMN1. Publikacja: 20 kwietnia 2016 r. Data udostępnienia: 25 kwietnia 2016 r. 9. Online Mendelian Inheritance in Man. Neuronopathy, distal hereditary motor, type VA; HMN5A. http://www.omim.org/entry/600794. Ostatnia edycja: 2 stycznia 2014 r. Data udostępnienia: 22 kwietnia 2016 r. 10. Irobi J, Dierick I, Jordanova A, Claeys KG, De Jonghe P, Timmerman V. Unraveling the genetics of distal hereditary motor neuropathies. Neuromolecular Med. 2006;8(1-2):131–146. 11. Glanzman AM, Mazzone E, Main M, et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20(3):155–161. 12. Romeo DM, Ricci D, Brogna C, Mercuri E. Use of the Hammersmith Infant Neurological Examination in infants with cerebral palsy: a critical review of the literature. Dev Med Child Neurol. 2016;58(3):240–245. 13. Mercuri E, Finkel R, Montes J, et al. Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul Disord. 2016;26(2):123–131. 14. Spinal Muscular Atrophy Clinical Research Center. Arkusz wyników badania CHOP INTEND dla SMA typu I. http://columbiasma.org/links.html. Data aktualizacji: 14 marca 2013 r. Data udostępnienia: 26 kwietnia 2016 r. 15. Dane własne. Biogen Inc, Cambridge, MA. 16. The Pediatric Neuromuscular Clinical Research Network for SMA. Expanded Hammersmith Functional Motor Scale for SMA (HFMSE). http://columbiasma.org/links.html. 7 marca 2009 r. Data udostępnienia: 25 kwietnia 2016 r. 17. Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704–712. 18. Arnold WD, Sheth KA, Wier CG, et al. Electrophysiological motor unit number estimation (MUNE) measuring compound muscle action potential (CMAP) in mouse hindlimb muscles. J Vis Exp. 2015;103:1–8. 19. Bromberg MB & Swoboda KJ. Motor unit number estimation in infants and children with spinal muscular atrophy. Muscle Nerve. 2002;25(3):445–447. 20. Finkel RS. Electrophysiological and motor function scale association in a pre-symptomatic infant with spinal muscular atrophy type I. Neuromuscul Disord. 2013;23(2):112–115.
W jaki sposób podstawowe czynności życiowe i postęp choroby dziecka z SMA wpływają na jego codzienną aktywność i życie?
Dowiedz się więcej